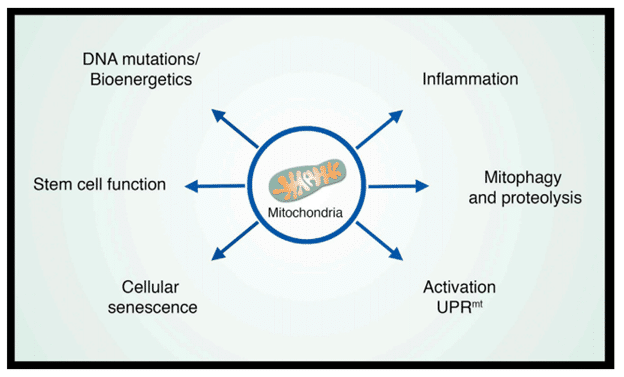
Mitochondria – More than Powerplants in Cells
Mitochondria are the large organelles that exist in most human cells. They are the critical organelles that generate ATP through the respiratory chain and produce the intermediate metabolites that are essential to various cellular processes. During aging, the respiratory chain losses its efficacy and the membrane of the mitochondria become leaky, which cause multiple aging phenotypes. In fact, aging research in past decades has suggested that mitochondria are the central node that are cross-talking with various age-related pathways. Therefore, mitochondria can no longer be viewed as simple power plants but rather as platforms for intracellular signaling, regulators of innate immunity, and modulators of stem cell activity. In turn, each of these properties provides clues as to how mitochondria might regulate aging and age-related diseases (Sun et al., 2016).
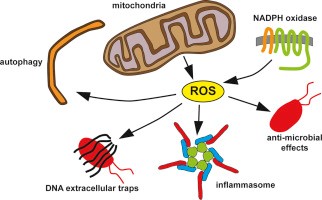
ROS and Oxidative Damage – Not the Primary Culprit?
When talking about mitochondria and aging, the mitochondrial free radical theory is one of several theories of aging that have been developed for decades. During the aging process, the dysfunctional mitochondria increase the production of reactive oxygen species (ROS). This highly reactive agent could cause oxidative damage, which further damage the mitochondria as well as the whole cell.
However, the role of ROS in aging has become controversial in recent years as some studies suggest that increased mitochondrial ROS and oxidative damage may prolong lifespan in yeast and C. ele-gans (Dunn et al., 2015). Moreover, genetic manipulations in mice that increase mitochondrial ROS and oxidative damage do not accelerate aging (see “Mice deficient in both Mn superoxide dismutase and glutathione peroxidase-1 have increased oxidative damage and a greater incidence of pathology but no reduction in longevity”). More importantly, mice with increased antioxidant defenses do not present an extended lifespan (see “The overexpression of major antioxi-dant enzymes does not extend the lifespan of mice”). Finally, various studies also point out that genetic manipulations that impair mitochondrial function, but do not increase ROS, accelerate aging.
This evidence suggests that the role of mitochondrial dysfunction in aging is far more complicated than oxidative damage caused by ROS.
Mitochondrial Biogenesis is Reduced During Aging
Besides ROS, mitochondrial dysfunction can contribute to aging through numerous mechanisms. One of them is the reduced biogenesis of mitochondria, or the decreased ability to generate new, healthy mitochondria. This can lead to various age-related physiological declines, including the deterioration of muscle function. Interestingly, physical exercise and dietary restriction (i.e., fasting) can alleviate mitochondrial degeneration and thus improve healthspan.
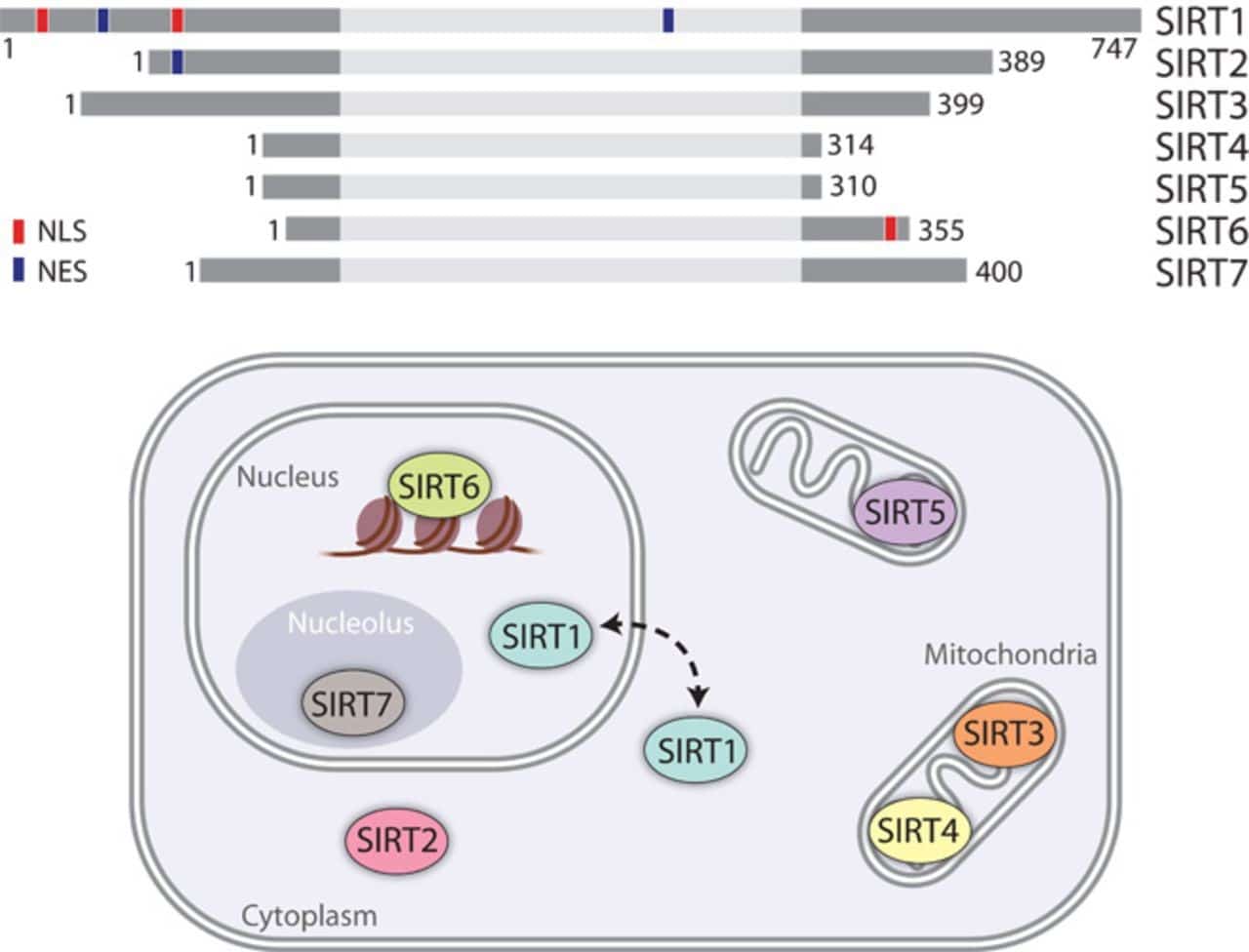
Sirtuins Regulate Mitochondral Biogenesis
There are seven Sirtuins in humans (SIRT1-7) which are the primary “longevity factors” that depend on NAD+. Interestingly, three of these seven proteins (SIRT3-5) are targeted to the mitochondria and SIRT1 itself is a regulator of mitochondrial biogenesis.
SIRT1 modulates mitochondrial biogenesis through a process involving the transcriptional coactivator, which activates a specific gene expression program and the removal of damaged mitochondria by autophagy (see “A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy”).
Besides SIRT1, the main mitochondrial deacetylase, SIRT3, can target many enzymes involved in energy metabolism, including respiratory chain and ketogenesis components. SIRT3 can also directly control the rate of ROS production by modifying the key enzyme in the process.
These results collectively suggest that sirtuins and NAD+ may control mitochondrial function and thus play a protective role against age-associated diseases.
Mitochondria, a Nexus for calorie restriction, NAD+ and Sirtuins
One of the most well-known mechanisms for longevity is calorie restriction. Surprisingly, calorie restriction-induced longevity is mediated (at least partially) by improved mitochondrial function and sirtuin activities.
In yeast, calorie restriction increased the mitochondrial respiration and activity of yeast SIR2 and thereby extended lifespan. In C. elegans, calorie restriction was shown to require two sensory neurons, which trigger an increase in mitochondrial respiration. In mice, calorie restriction is mediated by SIRT1, which increases respiration as well as the number of mitochondria per cell. NAD/NADH ratio and SIRT1 protein levels are increased in several rodent tissues during calorie restriction.
Finally, some of these effects found in mice also have been observed in a six-month human trial in which subjects were calorie restricted. Most strikingly, muscle punch biopsies from these calorie-restricted individuals showed upregulation of SIRT1, the mitochondrial protein TFAM, and the number of mitochondria compared to biopsies from control individuals on a regular diet.

These findings suggest that a conserved pathway may operate in mammals during calorie restriction involving the activation of SIRT1 and increased NAD+ that triggers an increase in mitochondrial activity, which in turn suggest the essential role of NAD+ in this process (read more at “Mitochondria—A Nexus for Aging, Calorie Restriction, and Sirtuins?”).